



Andrew Legan is a postdoctoral researcher at the USDA-ARS and University of Arizona in Tucson, AZ. He is a skilled science communicator and can be found on TikTok, Twitter, Instagram, and his personal website. Reach out to him on any of these platforms, or via email at andrew.w.legan[at]gmail[dot]com.
Overview
Gene duplication, redundancy, and divergence gives rise to groups of related genes with varied functions. When such multigene families scaffold the genetic architecture of a phenotype of interest, it’s important to account for all the constituent genes and to predict their gene models accurately to get a complete picture of how and why they contribute to higher-order phenotypes.
However, annotating multigene families is a challenging endeavor, especially in non-model organisms. Divergent sequence evolution in multigene families makes it difficult to accurately predict gene models.
This post briefly outlines a methodological roadmap to annotating gene families in non-model organisms, tailored by my experiences annotating insect odorant receptor genes. Be sure to also check out the accompanying interview with Dr. David Stern, whose team at Janelia Research Campus recently discovered a novel family of candidate effector genes in aphids, phylloxerids, and coccids (Korgaonkar et al. 2021; Stern and Han 2022).
1.) Ab initio gene prediction:
While ab initio gene annotation tools have the potential to accurately annotate most genes in the genome under the right circumstances, even very complete genome assemblies with ab initio annotations scoring in the top percentiles of BUSCO analyses may lack gene models for duplicated and highly divergent genes. Still, ab initio gene predictions are often accurate for conserved members of multigene families, and they may identify fragments of divergent genes.
2.) Basic local alignment search tool (BLAST)
Basic local alignment search tool (BLAST) is useful in searches for duplicated genes. Loci containing rapidly evolving duplicates in the insect odorant receptor gene family were uncovered in social insect genomes by using sequences of odorant receptors from the fly Drosophila melanogaster and honey bee Apis mellifera as queries in BLAST searches (Zhou et al. 2015).
Because amino acid sequences are more conserved than DNA sequences, protein sequences should be used as queries to search translated genomes with the TBLASTN algorithm. By increasing the E-value threshold to permit hits between more dissimilar query and target sequences, TBLASTN can identify portions of rapidly evolving duplicate genes that would have been overlooked. Using “blast2gff.py” the blast output table can be converted to a gff file and imported to a genome browser. Keep in mind that relaxing the E-value cutoff may generate misleading hits to conserved protein motifs in genes outside of the multigene family.
3.) Tissue-specific RNA sequencing:
What better information for identifying exon and intron boundaries than transcribed and processed messenger RNA? Paired-end and long-read RNA sequencing are especially useful for gene annotation. Align the mRNA reads to the genome and import alignments to a genome browser to guide manual annotation. Antennal mRNA-sequencing revealed many odorant receptor genes in an expanded and divergent “9-exon” clade in social wasp genomes (Figure 1).
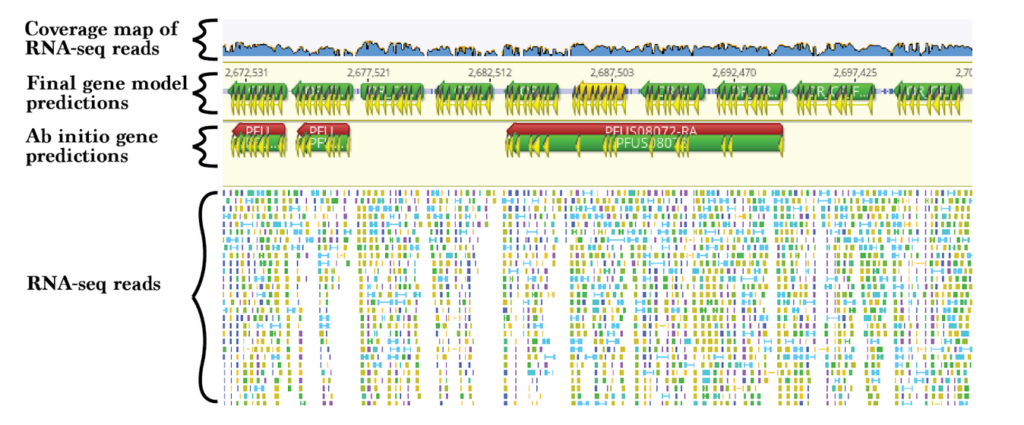
4.) Gene structure homology:
While rapidly evolving genes may be too divergent for sequence homology to be useful, the exon-intron structures of genes tend to be more conserved. This is illustrated by the odorant receptor genes in Figure 1. This clade is called the “9-exon” subfamily because each has nine exons, and these exons are of stereotypical lengths. The conserved nature of homologous gene structure was leveraged by Drs. Clair Han and David Stern in their search for gall effector genes of the Hemipteran bicycle gene family. Using a custom logistic regression classifier informed by gene structure homology, they uncovered many putative bicycle genes in aphids, phylloxerids, and coccids (Stern and Han 2022).
5.) Outcome:
The last step is combining all these lines of evidence to design accurate gene models. When deciding which evidence to prioritize in determining a particular gene structure, the March 2016 guidelines by the Human and Vertebrate Analysis and Annotation group are helpful, and many guidelines like canonical splice sites, start and stop codons apply to invertebrates as well.
In my opinion, the best approach to multigene family annotation integrates genomic and transcriptomic evidence which, while potentially expensive, permits accurate annotation of multigene families and creates new opportunities for research on non-model organisms.
References
Korgaonkar, A., Han, C., Lemire, A. L., Siwanowicz, I., Bennouna, D., Kopec, R. E., Andolfatto, P., Shigenobu, S., & Stern, D. L. (2021). A novel family of secreted insect proteins linked to plant gall development. Current Biology, 31(9), 1836-1849.
Legan, A. W., Jernigan, C. M., Miller, S. E., Fuchs, M. F., & Sheehan, M. J. (2021). Expansion and accelerated evolution of 9-exon odorant receptors in Polistes paper wasps. Molecular biology and evolution, 38(9), 3832-3846.
Stern, D. L., & Han, C. (2022). Gene structure-based homology search identifies highly divergent putative effector gene family. Genome biology and evolution, 14(6), evac069.
Zhou, X., Rokas, A., Berger, S. L., Liebig, J., Ray, A., & Zwiebel, L. J. (2015). Chemoreceptor evolution in Hymenoptera and its implications for the evolution of eusociality. Genome biology and evolution, 7(8), 2407-2416.