



About the Blog Author: Sheela Turbek is a postdoctoral researcher in Dr. Kristen Ruegg’s lab at Colorado State University. Her graduate research at the University of Colorado Boulder focused on how behavioral and phenotypic traits mediate patterns of genetic exchange between closely related taxa. Lately, however, she is interested in using genomic tools to advance the conservation of threatened avian species.
Genetic recombination – the process by which DNA strands are exchanged to produce new nucleotide combinations – is likely something you don’t think about every day. However, a quick glance in the mirror is enough to remind you of both its prevalence and importance in sexually reproducing organisms: recombination is the very reason you possess a unique blend of traits that distinguishes you from your parents. By generating novel genetic combinations upon which natural selection can act in each generation, this process plays a fundamental role in the evolution of populations.
Interestingly, the rate at which recombination occurs can vary widely between species and among regions within an individual’s genome (Stapley et al. 2017). Some genomic regions, known as recombination hotspots, shuffle genetic variation at extremely high rates, while other areas (e.g., centromeres) are essentially devoid of recombination. This variation in recombination rate across the genome can greatly influence estimates of genomic differentiation. In particular, FST estimates tend to be higher in genomic regions of low recombination, where the reduction in diversity due to linked selection extends over large genomic regions (Renaut et al. 2013).

Historically, variation in recombination rate and its impact on patterns of genomic differentiation was notoriously difficult to study, particularly in non-model organisms. However, with recent advances in high-throughput genomic sequencing methods, it is now possible to estimate fine-scale patterns of recombination from genomic sequencing data alone. In our recent study (Turbek et al. 2021a), funded through EECG awards from the AGA, we took advantage of emerging methods for estimating recombination rate to investigate the association between patterns of recombination and landscapes of genomic differentiation during the earliest stages of divergence. Many recently formed species are only distinguished by a few genomic regions that are involved in reproductive isolation and therefore highly differentiated relative to an otherwise identical genomic background (Martin et al. 2013; Figure 1). However, we still don’t know whether these divergence peaks that appear to be involved in species differences are associated with high, low, or average recombination rates in the earliest stages of the speciation process.
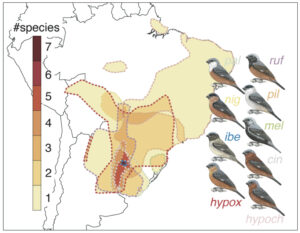
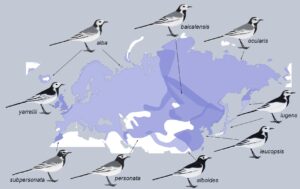
To address this question, we used published whole-genome sequencing data to generate fine-scale recombination maps from two avian radiations – southern capuchino seedeaters (Sporophila; Figure 2) and White Wagtails (Motacilla alba; Figure 3) – which have recently diversified in mate preferences and genes encoding plumage coloration to form a variety of different taxa (Semenov et al. 2021; Turbek et al. 2021b). Given their recent origin, the species and subspecies within these radiations are largely genetically identical; however, they exhibit a few narrow regions of elevated genomic differentiation that are involved in plumage patterning (Campagna et al. 2017; Semenov et al. 2021). We compared the patterns observed in these incipient species to those documented in Collared and Pied Flycatchers, more distantly related taxa that exhibit peaks and troughs of genomic differentiation formed in part by background selection (i.e., negative selection against deleterious alleles) (Burri et al. 2015).
On the one hand, genetic recombination breaks up combinations of alleles, suggesting that regions of elevated genomic differentiation may be found in areas of reduced recombination early in divergence, allowing diversification to proceed despite gene flow. On the other hand, however, recombination generates new genetic variation upon which natural selection can act. Therefore, divergence peaks containing pigmentation genes could alternatively be found in high recombination regions, which would help explain the astounding diversity of plumage patterns in both capuchinos and wagtails.
By comparing variation in recombination rate in each system, we found that patterns of recombination were highly similar among species within capuchinos, wagtails, and flycatchers. In spite of these largely conserved recombination rates, however, not all capuchino species exhibited the same divergence peaks, indicating that regions of elevated differentiation have not been generated by variation in recombination rate alone. We found a much weaker negative relationship between FST and recombination rate in capuchinos and wagtails than in Pied and Collared Flycatchers, suggesting that background selection has played a limited role in generating divergence peaks early in the speciation process. Nonetheless, all divergence peaks between capuchinos, wagtails, and flycatchers fell in regions on the lower end of the recombination spectrum. Given that recombination breaks up co-adapted combinations of genes, the location of divergence peaks underlying plumage differences in areas of low recombination likely protected these regions from the effects of gene flow, facilitating diversification in these rapid radiations. More broadly, our study highlights the importance of considering variation in local recombination rate when interpreting patterns of elevated genomic differentiation at every stage of the speciation process.
References