


**The AGA grants EECG Research Awards each year to graduate and post-doctoral researchers who are at a critical point in their research, where additional funds would allow them to conclude their research project and prepare it for publication. EECG awardees also get the opportunity to hone their science communication and write three posts over their grant tenure for the AGA Blog. In the first in the series, our EECG awardees write about their research and their interests as an ’embarkation’.

About the author: Matthew Gibson (he/him) is a PhD candidate in Dr. Leonie Moyle’s lab at Indiana University Bloomington. His research focuses on understanding the role of abiotic climate in shaping patterns of adaptive genomic variation in wild tomato species. To do so, he uses a combination of computational and population genetic approaches.
Because they are sessile, wild plants are forced to cope with unpredictable and harsh environmental conditions. As a result, natural plant populations are often subject to strong natural selection favoring traits that suit them to their local environment—a process termed local adaptation. Although nearly a century of experiments has revealed that local adaptation is pervasive in plant populations sourced from disparate habitats, our understanding of the processof local adaptation at the genetic level remains limited. Key questions are whether adaptation usually proceeds from standing variation or de novo population-specific changes and, more generally, whether adaptation to similar environments is predictable. The answers to these questions have broad implications for our understanding of evolution and its applications, including how variation is maintained in natural populations, whether evolutionary outcomes of climate change can be predicted, and whether naturally selected alleles can be harnessed for crop breeding.
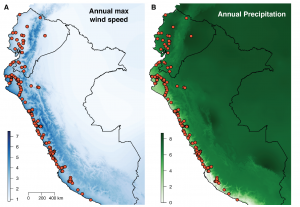
My research addresses these questions in an ecologically diverse species of tomato—Solanum pimpinellifolium—using several computational approaches. Myself and others have shown that climate and other abiotic factors play a fundamental role in structuring genomic variation in this species (Figure 1; Figure 2B; Nakazato et al., 2008; Gibson & Moyle, 2020). In particular, an abiotic gradient extending from low elevation coastal sites (dry) to high elevation montane sites (wet) in Ecuador is thought to impose strong selection on traits related to drought and/or temperature tolerance.
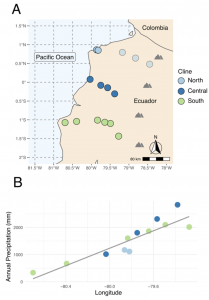
My most recent project aims to leverage a replicated population sampling scheme—densely sampled coastal-inland transects from three latitudes (Figure 2A)—to determine whether climate adaptation in this species is associated with parallel vs unique genetic changes, and whether adaptation to these habitats proceeded via de novo mutations or was co-opted from standing variation. To address this requires identification of shared and unique genomic signals of coastal-inland adaptation along the three gradients as well as inference of their evolutionary origins.
By generating genome sequences from populations situated along the three replicate gradients and combining them with publicly available climate data, it is possible to detect alleles subject to selection by investigating spatial associations of allele frequencies with specific climate variables using statistical techniques similar to genome wide association studies (GWAS). By then comparing these associations between replicate gradients, I can identify alleles that are shared (i.e., targeted by selection multiple times, either independently or non-independently) and unique (i.e., only selected along a single gradient). I can then leverage population genetic theory (Lee & Coop, 2017) to determine whether candidate alleles putatively under selection along multiple gradients were each derived de novo(i.e., convergent evolution) or from existing pools of variation. This is possible because past selection should leave behind characteristic signals (e.g., reductions in nucleotide diversity around selective sites indicative of selective sweeps) and/or patterns of haplotype sharing between replicate gradients that can be used to infer the most likely source of the adaptive allele. Together these results will characterize the origins of adaptive genetic variation in the ecologically diverse (and agronomically important) species S. pimpinellifolium, as well as speak to the predictability of adaptive evolution more generally.
References