


**The AGA grants EECG Research Awards each year to graduate students and post-doctoral researchers who are at a critical point in their research, where additional funds would allow them to conclude their research project and prepare it for publication. EECG awardees also get the opportunity to hone their science communication and write posts over their grant tenure for the AGA Blog. In the first in the series, our EECG awardees write about their research and their interests as an ’embarkation’.**


About the blog authors: Karina (she/her) and Mark (he/him) (see the lab website) are postdoctoral researchers within the Illera lab at the University of Oviedo working on the evolutionary mechanisms driving gut microbiome biogeography.
Until recently, microbial taxa were largely considered cosmopolitan. Deep sequencing of microbial communities in diverse environments has revealed a more complex scenario, where dispersal is limited by suitable habitats and interspecific competition (Martiny et al. 2011). In bacterial gut microbiomes, both host selection and high competition may restrict dispersal and drive biogeographical patterns (Snijders et al. 2017; Verster and Borenstein 2018). Gut bacterial communities often show congruent evolutionary histories with their hosts, which has led to the expectation that co-evolution is the main mechanism driving this pattern. This issue remains contentious, since distinguishing co-evolution from vicariance (due to selective pressure from shared biogeographical patterns) is challenging (Groussin et al. 2020).
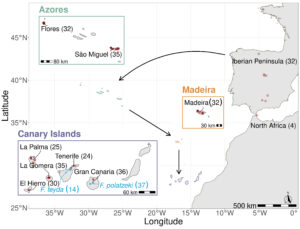
As a model host system, we are using the colonization history (dated between 0.1-1 Ma) and the resolved phylogeny of the the common chaffinch species complex (Fringilla spp) in Macaronesia and neighboring mainland regions (Iberian Peninsula and North Africa) (Figure 1.) (Recuerda et al. 2021). Moreover, our study system provides an opportunity to disentangle between host relatedness and distribution patterns by incorporating the sympatric blue finches, two sister species (diverged ~ 2 Ma from the common chaffinch species complex) endemic to the Canary Islands: F. teydea and F. polatzekii (Figure 1). In addition, we opportunistically sampled individuals from 14 passerine species (distributed across 10 Families) providing phylogenetic outgroups for our analyses.
Using oceanic island biogeography as a natural experiment with discrete, replicated units, we aim to: i) estimate the
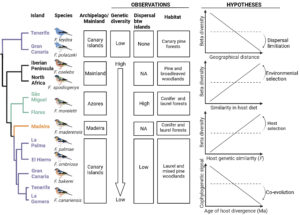
strength of geographic distance, diet, and host genetic similarity in shaping biogeographical patterns of gut bacterial assemblage; ii) assess the degree of phylogenetic congruence between host and gut bacterial communities; iii) test for patterns of cophylogenetic signals between specific core bacterial lineages, functional groups, and host diversification events, and; iv) identify host genomic targets of bacterial selection and their association to cophylogeny (Figure 2).
So far, studies elucidating the mechanisms driving congruent evolutionary histories between the host and gut microbiomes have resulted in conflicting results, particularly in birds (Capunitan et al. 2020). This likely stems from the difficulty of controlling for the complex web of ecological determinants (Groussin et al. 2020) and the coarse grain of the taxonomic scale of short-read 16S profiling (Ladau and Eloe-Fadrosh 2019). We address this issue by: i) using a replicated island system, ii) accounting for the effect of diet using DNA metabarcoding (rcbL marker for plants and COI for invertebrates), iii) using high bacterial taxonomic resolution by means of synthetic long-read sequencing of the whole 16S region (1500 bp of the 16S region; LoopSeq technology).
With the support of the EECG Research Award, we plan to conduct genome wide profiling of Fringilla (through genome-by-sequencing) to take into account host whole genome effects on microbial diversity and community structure. The inclusion of host genetics, a crucial component of gut microbiomes, will enable us to move beyond biogeographical patterns and disentangle in a holistic manner the mechanisms driving them.
References: