


**The AGA grants EECG Research Awards each year to graduate students and post-doctoral researchers who are at a critical point in their research, where additional funds would allow them to conclude their research project and prepare it for publication. EECG awardees also get the opportunity to hone their science communication and write posts over their grant tenure for the AGA Blog. In the first in the series, our EECG awardees write about their research and their interests as an ’embarkation’.**

About the author: Daniel Shaw is a PhD candidate at the University of Georgia working with Dr. Michael White. Daniel is interested in understanding how evolutionary processes shape genome evolution and gene regulation. His PhD is focused on the evolution of gene regulation on X and Y chromosomes in stickleback fish. Keep up with Daniel on twitter @dna_shaw.
Biological sex is diverse across the tree of life (Bachtrog et al., 2014). Many people are familiar with the X and Y chromosomes that contribute to sex determination in mammals1. Outside of mammals, there are many ways to create sexual dimorphic individuals. It remains unclear how diverse mechanisms of sex determination evolve. One way to address this question is to study the evolution of sex chromosomes.
Ancestrally, most X and Y chromosomes evolved from autosomal pairs. As autosomes, they freely exchanged DNA through meiotic recombination. This process helps chromosomes remain genetically similar. The evolution of distinct sex chromosomes (X and Y) is due to the suppression of recombination. Without recombination, the X and Y chromosome will start to accumulate their own set of mutations, resulting in genetic differences between the two. The X chromosome still undergoes recombination in XX individuals. But the loss of recombination is much more drastic on Y chromosomes. Y chromosomes essentially evolve as one large linkage group. This makes selection less efficient. Without crossovers to separate haplotypes, selection must act on all of the alleles on the Y chromosome, rather than separating them (Bachtrog, 2013). This means that most Y chromosomes have been force to carry the good, the bad, and the ugly! It seems that the only way to rid Y chromosomes of bad alleles is to degenerate and lose genes entirely. This is likely why older Y chromosomes (like mammals) consist of very few genes and appear much smaller than X chromosomes.

In our lab, we focus on the threespine stickleback fish (Gasterosteus aculeatus) (Figure 1). Across all fish, sex determination varies greatly. But multiple species of stickleback have evolved sex chromosomes within the last 30 million years (Ross et al., 2009). While 30 million years may seem like a pretty long time, we consider this “recent” compared to the ~200-million-year-old mammalian Y. On this slightly smaller time scale, the threespine stickleback Y chromosome still contains about half the genes as the X chromosome. Many of these genes are still functional but have accumulated a lot of unique mutations. We can use the stickleback Y chromosome to understand more about the early steps of Y evolution and degeneration.
In addition to identify how sex chromosomes evolve, studying recently evolved sex chromosomes can help us
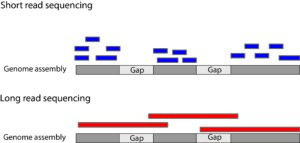
understand fundamental processes related to genetics and evolution. One example of this is allele specific expression. Where two similar copies of the same gene are expressed differently. Allele specific expression has been identified across the genome. However, this process is difficult to study and we do not have a deep understanding of how allele specific expression evolves. In the case of the stickleback X and Y chromosome, we have an excellent model to study this effect. While there are 500 genes still shared between the X and Y, they are expressed at different rates when quantified by RNA sequencing (Peichel et al., 2020; White et al., 2015). This means that since the X and Y started evolving, they have been accumulating mutations that alter the expression patterns of the shared genes. Over the course of my PhD, I have been using high quality genome assemblies to identify regions of DNA that regulate these genes. When we sequence extra-long pieces of DNA we can build assemblies that include very repetitive sequence like that found on the Y chromosome (Nath et al., 2021; Peichel et al., 2020) (Figure 2). We can compare the DNA sequence between the X and the Y to identify unique mutations. Using this assembly, we can identify important regions in intergenic space that likely regulate neighboring genes. By finding these putative regulatory elements on the stickleback Y chromosome, I have started to figure out how allele specific expression can evolve so rapidly.
Recently, I was awarded an EECG grant from AGA. We plan to use this funding to generate ultra long read sequencing on the Oxford Nanopore platform. By sequencing long stretches of DNA, we will be able to assemble highly repetitive regions We will be able to extend our comparative analyses to newly assembled regions to study structural variation between the X and Y chromosome. I am particularly excited to compare evolutionary rates between the X and Y centromeric repeats. I hope this data set will enable us to learn more about how sex chromosomes evolve quickly after the suppression of recombination and perhaps teach us about more fundamental processes in evolutionary genetics.
Footnote:
1.Here, I refer to biological sex as a trait distinct from gender. Within species, like humans, sex is often more complicated than the way it presented in scientific and popular media. In humans, sexual characteristics are derived from a mix of DNA, hormones, and anatomy development within individuals.
References